This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison, Spring 2012.
SOD1 Homologs and Alignments

What is a homolog?
A homolog is a gene in one organism that is related to a gene in another organism by descent from a common ancestor. Therefore, homologs can help us to better understand similarities and differences between organisms on the genetic level. A gene that is well conserved likely has an important function in multiple organisms. Because of the redunancy of the genetic code, homologs are expected to be less similar in DNA base pairs than amino acids.
Identifying homologs is a critical step in the study of human diseases, as the early research to understand and treat diseases like ALS is typically done using model organisms.
Identifying homologs is a critical step in the study of human diseases, as the early research to understand and treat diseases like ALS is typically done using model organisms.
The SOD1 gene is conserved in chimpanzee, dog, cow, mouse, rat, chicken, zebrafish, fruit fly, mosquito, nematodes, S.pombe, yeast, K.lactis, E.gossypii, M.grisea, N.crassa, A.thaliana, and rice.
Gene Sequences
Below are links to the DNA sequences of SOD1 homologs in selected model organisms:
|
Pan troglodytes (Chimpanzee) superoxide dismutase 1 (Sod1): NC_006488.2 sequence length: 7,444 bp Mus musculus (Mouse) superoxide dismutase 1 (Sod1): NC_000082.5 sequence length: 5,583 bp Rattus norvegicus (Rat) superoxide dismutase 1 (Sod1): NC_005110.2 sequence length: 5,577 bp Danio rerio (Zebrafish) superoxide dismutase (Sod): NC_007121.5 sequence length: 5,035 bp |
Drosophila melanogaster (Fruit Fly) superoxide dismutase (SOD): NT_037436.3 sequence length: 1,458 bp Caenorhabditis elegans (Roundworm) superoxide dismutase 1 (SOD-1): NC_003280.8 sequence length: 1,181 bp Saccharomyces cerevisiae (Yeast) sod1p (SOD1): NC_001142.9 sequence length: 465 bp Arabidopsis thaliana (Mouse-ear Cress) superoxide dismutase (CSD1): NC_003070.9 sequence length: 2,255 bp |
BLAST Alignments
The following table shows the results of the Nucleotide BLAST alignment of Human SOD1 to the homolog of the eight organisms above [1]. The alignment was created using Megablast, searching for highly similar sequences. Organisms not included in the table did not have enough DNA sequence similarity to form a significant alignment.

Below is the BLAST alignment of the same sequences using blastn, searching for somewhat similar sequences [2]. blastn was able to create a significant alignment for each of the model organisms used.
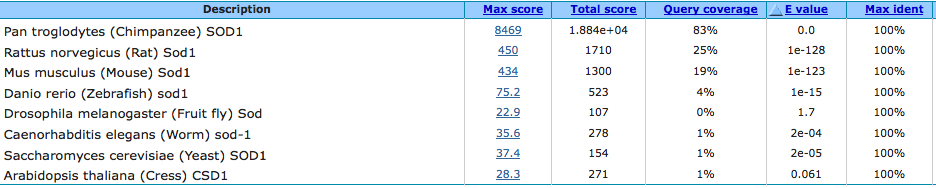
These alignments are ordered by E value of the closest single alignment, which is a measure of similarity in which 0 is the most similar and higher numbers are less similar. For NCBI News' explanation of the other metrics displayed, click here.
T-Coffee Alignment
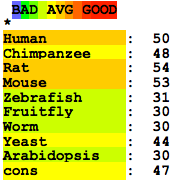
This file shows the T-Coffee alignment of the human SOD1 gene sequence to the eight organisms listed above using the default settings [3,4]. Below is the summary of the predicted accuracy of this alignment. The average predicted accuracy is due to the vastly different intronic sequences in the homologs. This uncertainty contrasts the protein alignment.
Analysis
Because the human SOD1 gene has a relatively short coding region compared to its non-coding region, DNA sequence variability in the non-coding regions is expected to arise between species throughout evolution. At 9,310 base pairs, the human SOD1 gene has significantly longer introns than many of its homologs. These differences account for much of the variability between species and the inability of BLAST to align the majority of the DNA sequences between humans and the most distantly related model organisms (query coverage of 0-1%). These differences can be clearly visualized in the T-Coffee alignment in the stretches where the human SOD1 sequence is not aligned to any of its homologs, and this leads to the uncertainty in the alignment seen in the summary. Despite such widely variable sequences, the proteins remain quite similar (see protein homology).
1. Zhang Z, Schwartz S, Wagner L, Miller W. (2000) A greedy algorithm for aligning DNA sequences.
J Comput Biol 7(1-2):203-14. PMID:10890397
2. Altschul S, Madden L, Schäffer A, Zhang J, Zhang Z, Miller W, Lipman D. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
PMID:9254694
3. Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39:W13-7. PMID:21558174
4. Notredame C, Higgins D, Heringa J. (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignments. JMB 302: 205-217. PMID:10964570
J Comput Biol 7(1-2):203-14. PMID:10890397
2. Altschul S, Madden L, Schäffer A, Zhang J, Zhang Z, Miller W, Lipman D. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
PMID:9254694
3. Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39:W13-7. PMID:21558174
4. Notredame C, Higgins D, Heringa J. (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignments. JMB 302: 205-217. PMID:10964570

{kind=link}