This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison, Spring 2012.
Phylogenetic Trees: SOD1 Protein

|

|

|

|
What is a phylogeny?
A phylogeny is a depiction of changing organismal lineages over time, where branch points represent a common ancestor of the two species. Using the degree of similarity between homologous protein sequences allows us to estimate how related species are to each other and to construct the phylogenetic trees seen below.
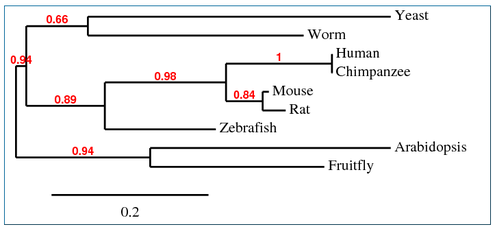
Phylogeny.fr Tree
The following alignment of Homo sapiens SOD1 with homologs in P. troglodytes, M. musculus,
R. norvegicus, D. rerio, D. melanogaster, A. thaliana, C. elegans and S. cerevisiae was generated by Phylogeny.fr using "one click" mode [1,2,3,4,5,6,7].
R. norvegicus, D. rerio, D. melanogaster, A. thaliana, C. elegans and S. cerevisiae was generated by Phylogeny.fr using "one click" mode [1,2,3,4,5,6,7].
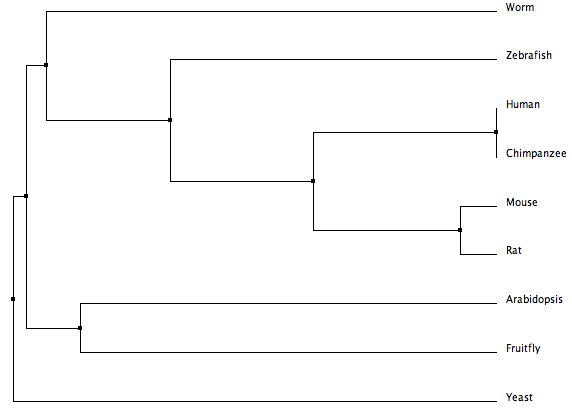
MUSCLE Phylogeny
The following tree was created by MUSCLE using default settings [3]. It was calculated using average distance based on percent identity.
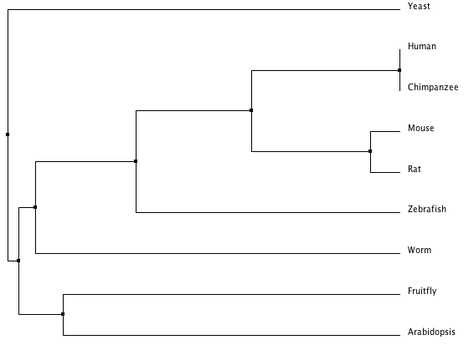
Clustal Omega Phylogeny
The following phylogeny was generated using Clustal Omega [8]. It was calculated using average distance based on percent identity.
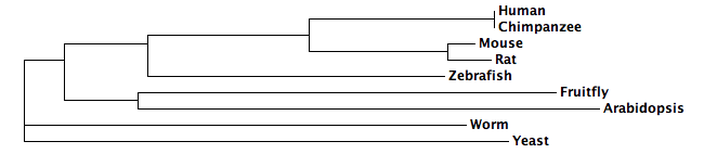
ClustalW Phylogeny
The following tree was created at ClustalW using default settings [9,10].
Analysis
The trees from Phylogeny.fr, Clustal Omega, ClustalW and MUSCLE are all fairly similar. Since the protein is well conserved and can be aligned with great accuracy (see protein homology), the different algorithms used by each tool come to similar conclusions. Phylogy.fr and MUSCLE differ by their placement of the branching of the C. elegans lineage. Since Phylogeny.fr uses MUSCLE alignment in it program, this change must come from the tool used to make the tree. Clustal Omega, an improvement on ClustalW, also moves the C. elegans lineage.
The Clustal programs are not consistently different from the MUSCLE alignments despite using different algorithms. While Clustal uses a standard progressive alignment method, comparing sequences two by two, MUSCLE uses iterative methods, further aligning subgroups to create a more accurate alignment [11]. They are equally user-friendly when accessed from EBI, so comparing a few different alignments may produce the best results.
The Clustal programs are not consistently different from the MUSCLE alignments despite using different algorithms. While Clustal uses a standard progressive alignment method, comparing sequences two by two, MUSCLE uses iterative methods, further aligning subgroups to create a more accurate alignment [11]. They are equally user-friendly when accessed from EBI, so comparing a few different alignments may produce the best results.
_____________________________________________________________________________________________________
- Dereeper A., Audic S., Claverie J., Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010 Jan 12;10:8. PMID:20067610
- Dereeper A.*, Guignon V.*, Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M., Claverie J.M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W465-9. Epub 2008 Apr 19. PMID:18424797
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, Mar 19;32(5):1792-7. PMID:15034147
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, Apr;17(4):540-52. PMID:10742046
- Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, Oct;52(5):696-704. PMID:14530136
- Anisimova M., Gascuel O. Approximate likelihood ratio test for branchs: A fast, accurate and powerful alternative. Syst Biol. 2006, Aug;55(4):539-52. PMID:16785212
- Chevenet F., Brun C., Banuls AL., Jacq B., Chisten R. TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics. 2006, Oct 10;7:439. PMID:17032440
- Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7. PMID:21988835
- Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R, Thompson J, Gibson T, Higgins D. (2007). Clustal W and Clustal X Version 2.0. Bioinformatics, 23, 2947-2948. PMID:17846036
- Thompson J, Higgins D, Gibson T. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673-4680. PMID:7984417
- Taly, J et al. (2011). Using the T-Coffee package to build multiple sequence alignments of protein, RNA, DNA sequences and 3D structures. Nature Protocols 6:1669-1682. PMID:21979275
