This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison, Spring 2012.
Conclusions
The goal of these experiments was to better understand how mutations in SOD1 lead to ALS. It is well documented that SOD1 mutants confer toxicity to neurons through abnormal gain-of-function processes, but the exact mechanism by which this occurs is unknown. I started my research by finding SOD1 homologs in model organisms. The SOD1 protein is well conserved in many organisms, including commonly used model organisms such as mice, zebrafish, nematodes, fruit flies and yeast, and I determined their evolutionary relationships by making phylogenetic trees. Because SOD1 is so well conserved, ALS models in these organisms, especially in mice, have been very useful in studying the disease.
In order to gain a better understanding of how mutant SOD1 might be causing ALS, I looked at the gene ontology to learn what is known about the cellular localization and molecular function of both wildtype and mutant SOD1 as well as the biological processes in which it is involved. I found two of the biological processes, apoptosis and mitochondrial respiration, intriguing because they are both mitochondrial-driven processes. Since SOD1 is normally located in the cytoplasm, it is likely that mutant SOD1 is affecting mitochondria. Looking at the known cellular pathways involved in ALS, I found that mutant SOD1 is affecting mitochondrial through aggregates, although the mechanism behind this process is unclear. I hypothesized that the aggregates in ALS motor neurons could be similar to aggregates in other neurodegenerative diseases, seen below:
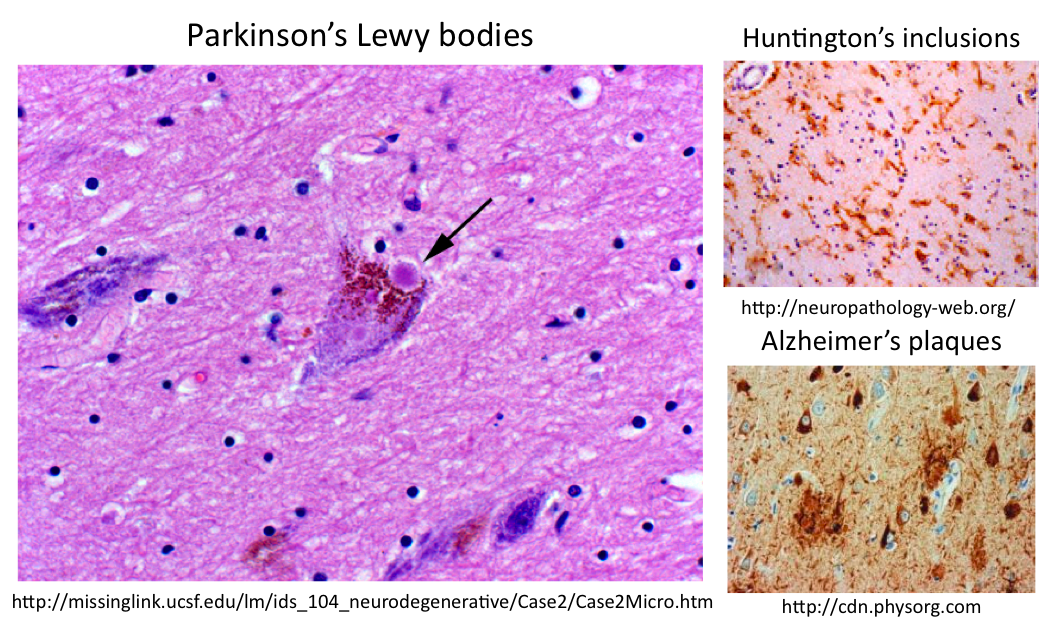
Using STRING interaction networks, I noticed that human SOD1 connects to Park7, a gene in which mutations cause Parkinson's disease. Parkinson's Lewy bodies are known to contain both ubiquitin and neurofilaments, so I looked at SOD1 interaction networks for these proteins or the mitochondrial proteins that may be affected by ALS aggregates. I found both ubiquitin-related proteins in SOD1 human and mouse interaction networks, and I discovered using gene ontology that about one half of all of the proteins in all of the model organism networks were mitochondrial proteins. These mitochondrial proteins include Bcl-2, an antiapoptotic protein, as well as ATP synthase, a protein critical for mitochondrial respiration, each linking back to the SOD1 mitochondrial biological processes. Therefore, I hypothesize that ALS aggregates may contain SOD1, ubiquitin, neurofilaments and potentially, mitochondrial proteins important to apoptosis or respiration.
In order to test this hypothesis, I proposed taking a proteomic approach to discovering the contents of ALS aggregates. I believe that zebrafish would be a good model organism with which to work as ALS models have been previously established, zebrafish neurons can be cultured for ease of use and zebrafish are a versatile organism in which mutants and transgenics can be quickly made in the continued study of ALS. Also, looking at the zebrafish SOD1 interaction network, I found that it, like the human SOD1 network, contains Park7, and the SOD1/Park7 combined network contains a ribosomal ubiquitin, suggesting that ALS aggregates in zebrafish may be similar to known neurodegenerative aggregates like Lewy bodies. By culturing SOD1 mutant zebrafish neurons, using subcellular fractionation to separate out the aggregates and then using MudPIT to identify the proteins, we could learn the contents of ALS aggregates. This knowledge could lead to new therapeutic targets or to new connections between ALS and other well-studied neurodegenerative diseases.
Since ALS aggregates may contain mitochondrial proteins leading to defects that cause ALS, I hypothesized that ALS-causing mutations in genes other than SOD1 may be localized to the mitochondria. While there are 38 different genes associated with ALS, only one of them encodes a mitochondrial protein: mitochondrial metalloendopeptidase OMA1, which is a negative regulator of mitochondrial fusion. Thinking back to my proposed aggregate contents, I realized that neurofilaments, the cytoskeletal proteins on which mitochondria move allowing for normal fusion and thus normal mitochondrial function, may be the key component in ALS aggregates that leads to mitochondrial defects. Therefore, I proposed the following model:
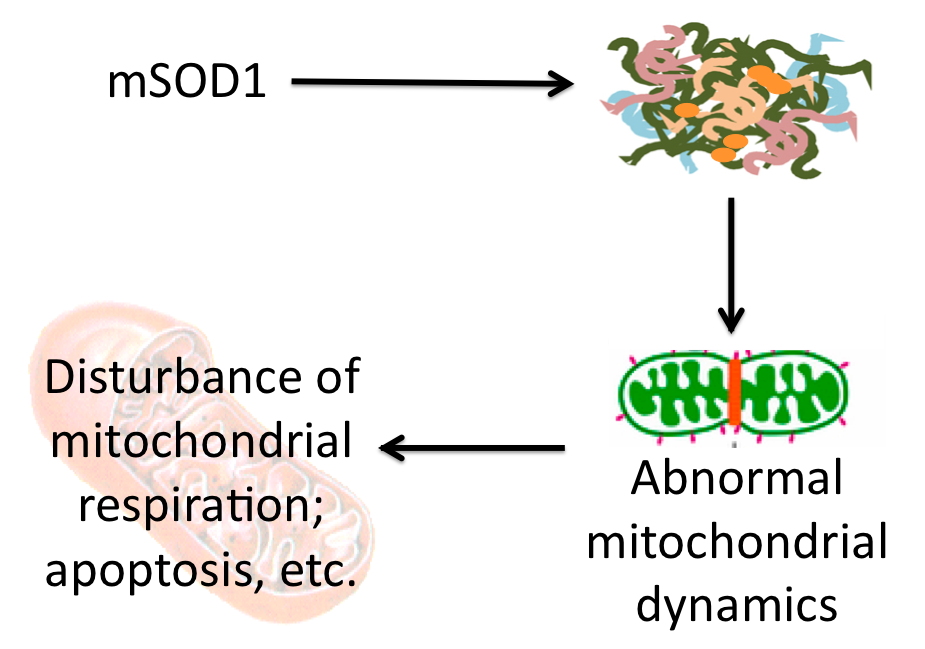
Proposed model of action by which aggregates may lead to mitochondrial defects in which aggregates containing neurofilaments disrupt mitochondrial dynamics, leading to dysfunction. (Click to enlarge.)
While this model leaves many questions unanswered, it may provide insight into how aggregates of misfolded proteins lead to neuron degeneration and could provide a link between disease progression in multiple neurodegenerative diseases as neurofilaments are common to aggregates across diseases. Alternatively, a recent advance in neurodegeneration research suggests that it could be the general accumulation of misfolded proteins in aggregates that leads to degeneration through alternate pathways not necessarily involving mitochondria [1]. Click here to read more about this fascinating advance in neurodegenerative therapy.
The continued study of ALS aggregates may help us to not only better understand the disease progression of ALS through mitochondrial defects or other mechanisms, but may also provide connections to other neurodegenerative diseases that could lead to broad advances in therapy.
Future Directions
In addition to the proposed proteomic experiment above and the study of aggregates in ALS, I proposed two future lines of inquiry to better understand the role of mitochondrial dynamics in ALS:
1. The continued characterization of OMA1, known to cause ALS by misregulation of mitochondrial fusion. This may include studying mitochondrial dynamics in OMA1 mutants and determining proteins that interact with OMA1.
2. A chemical genetics screen looking for potential therapies to help regulate mitochondrial fission and/or fusion.
1. The continued characterization of OMA1, known to cause ALS by misregulation of mitochondrial fusion. This may include studying mitochondrial dynamics in OMA1 mutants and determining proteins that interact with OMA1.
2. A chemical genetics screen looking for potential therapies to help regulate mitochondrial fission and/or fusion.
Below are the final copies of the presentation of this material in PowerPoint and PDF formats:
|
|
||||
1. Moreno J, Radford H, Peretti D, et al. (2012) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012. DOI: 10.1038/nature11058
