This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison, Spring 2012.
Amyotrophic Lateral Sclerosis (ALS)
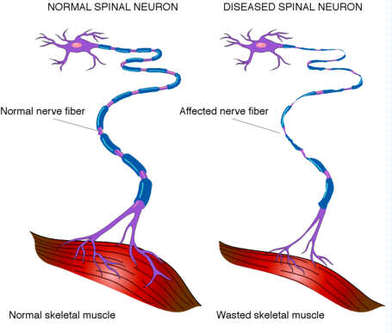
Neuronal degeneration in ALS.
http://www.neurotexasinstitute.com/
Amyotrophic Lateral Sclerosis (ALS), commonly referred to as Lou Gehrig's disease, is a neurological disease characterized by the degeneration of motor neurons. As motor neurons die, the brain loses the ability to send signals to muscles, which subsequently weaken, twitch and atrophy, eventually causing patients to lose their ability to move. Once muscles in the diaphragm and chest wall are affected, patients lose the ability to breathe, leading to respiratory failure. The disease is generally fatal within three to five years after the onset of symptoms, although about ten percent of patients live more than ten years with symptoms (NINDS).
While ALS may affect a patient's memory and decision making in addition to motor control, intelligence is usually unaffected along with the ability to see, hear, taste and touch. Thus, it is possible for patients to lead productive lives despite physical disability. Stephen Hawking, renowned physicist at the University of Cambridge, has lived over 40 years with ALS, contributing an astonishing amount to his field and popular science while suffering from the disease.
While ALS may affect a patient's memory and decision making in addition to motor control, intelligence is usually unaffected along with the ability to see, hear, taste and touch. Thus, it is possible for patients to lead productive lives despite physical disability. Stephen Hawking, renowned physicist at the University of Cambridge, has lived over 40 years with ALS, contributing an astonishing amount to his field and popular science while suffering from the disease.
ALS affects approximately two per every 100,000 people worldwide. In the United States, about 5,600 people are diagnosed with the disease each year, with about 30,000 Americans living with ALS at any given time. Anyone can get the disease as there are no clearly associated risk factors, and 90-95% of ALS cases are sporadic. The remaining 5-10% of cases are familial with about 20% of these inherited cases caused by mutations in the gene copper-zinc superoxide dismutase 1, commonly called SOD1 (ALSA).
SOD1
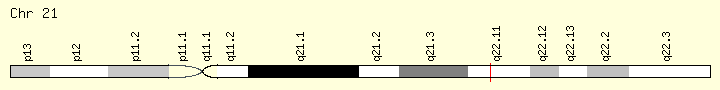
www.genecards.org
In humans, the SOD1 gene is located on chromosome 21 at location: 21q22.11. The 9,310 base pair gene has 5 exons and encodes a short 153 amino acid protein.
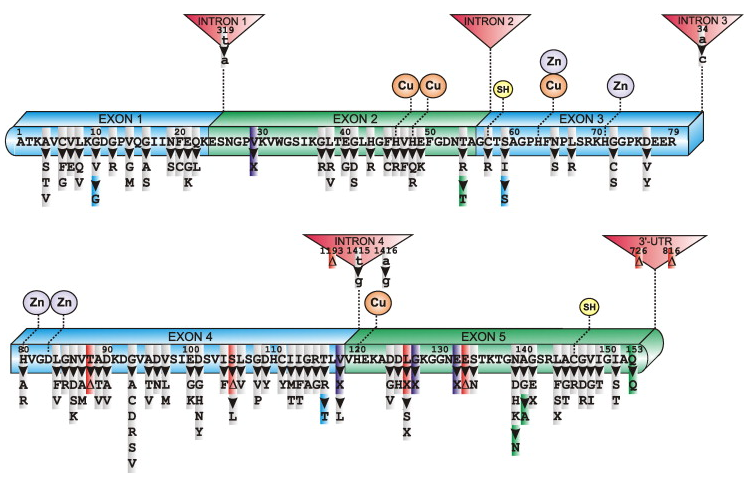
Human SOD1 primary sequence with exons, introns, Cu and Zn binding domains, intramolecular disulfide bond (SH) and mutations linked to sporadic and familial ALS. Mutation legend: grey, missense; purple, insertion; red, deletion; blue/green, silent; (Δ) inframe deletion; X, truncation [1]. (Click to enlarge.)
As many as 100 different mutations in SOD1 have been linked to familial ALS (ALSA). These mutations are scattered throughout the gene but have the highest prevalence in exons 4 and 5 [1]. An updated list of SOD1 mutations and mutations in other genes known to cause ALS can be found here.
When functioning normally, the SOD1 protein homodimerizes and binds zinc and copper ions, turning the products of oxidative phosphorylation into hydrogen peroxide and thus destroying harmful free superoxide radicals in the cytoplasm of cells (NCBI). These Zn and Cu binding sites are well conserved throughout evolution, as are the dimer interfaces (see protein homology). Researchers have hypothesized that mutant SOD1 takes on a toxic gain-of-function form, causing a wide range of cellular defects including mitochondrial dysfunctions, oxidative stress, calcium misregulation, aggregation of aberrantly processed proteins, endoplasmic reticulum (ER) stress, axonal transport disruption, neurotransmitter misregulation, programmed cell death and inflammation [2]. While genetic mutations in SOD1 cause familial ALS, defects in normal post-translational modifications can cause sporadic ALS in patients with wild-type SOD1 genes [3]. Therefore, the study of SOD1 can lead to a better understanding of the basis of both familial and sporadic ALS.
When functioning normally, the SOD1 protein homodimerizes and binds zinc and copper ions, turning the products of oxidative phosphorylation into hydrogen peroxide and thus destroying harmful free superoxide radicals in the cytoplasm of cells (NCBI). These Zn and Cu binding sites are well conserved throughout evolution, as are the dimer interfaces (see protein homology). Researchers have hypothesized that mutant SOD1 takes on a toxic gain-of-function form, causing a wide range of cellular defects including mitochondrial dysfunctions, oxidative stress, calcium misregulation, aggregation of aberrantly processed proteins, endoplasmic reticulum (ER) stress, axonal transport disruption, neurotransmitter misregulation, programmed cell death and inflammation [2]. While genetic mutations in SOD1 cause familial ALS, defects in normal post-translational modifications can cause sporadic ALS in patients with wild-type SOD1 genes [3]. Therefore, the study of SOD1 can lead to a better understanding of the basis of both familial and sporadic ALS.
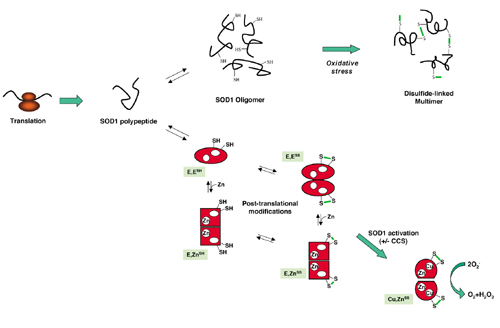
Where wild-type SOD1 dimerizes, acquires Zn ions and is activated by Cu ions, mutant SOD1 in ALS is prone to form disulfide-linked multimers under conditions of oxidative stress [4]. (Click to enlarge.)
Treatment for ALS
ALS is irreversible and thus difficult to treat. Drugs can be used to help slow the progression of symptoms and make those suffering from ALS more comfortable. Currently there is only one FDA approved treatment for patients with ALS, a drug called riluzole (Rilutek), which appears to slow the progression of the disease, possibly by reducing levels of glutamate in the brain, which are often elevated in patients with ALS (Mayo Clinic).
The recent progress in studying ALS and searching for treatments is astonishing, and the wealth of research brings hope that new treatments will be discovered. Check out the Clinical Trials page for more information on therapeutic targets and the clinical trials testing new treatments.
The recent progress in studying ALS and searching for treatments is astonishing, and the wealth of research brings hope that new treatments will be discovered. Check out the Clinical Trials page for more information on therapeutic targets and the clinical trials testing new treatments.
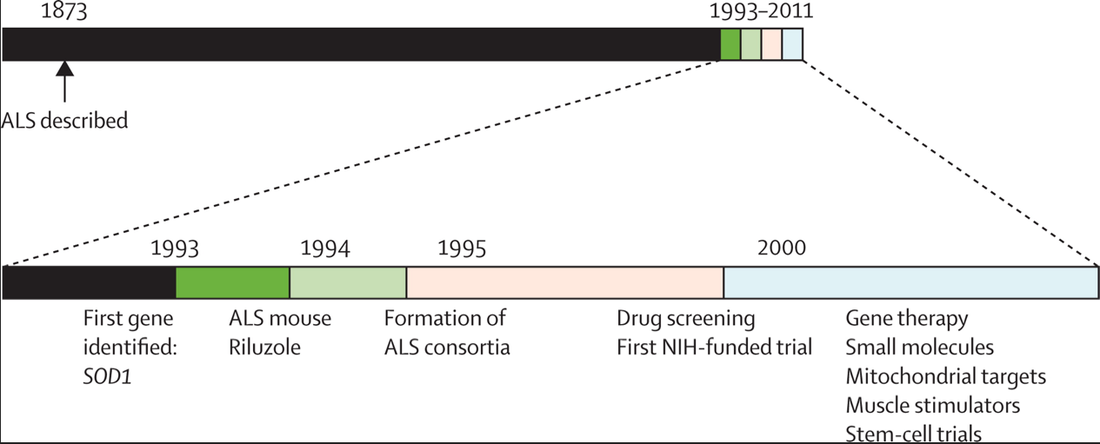
A timeline highlighting the increasing pace of ALS research [5].
1. Turner, B. and Talbot, K. (2008) Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Progress in Neurobiology 85 (1): 94-134. PMID:18282652
2. Faes, L. and Geert, C. (2011) Mitochondrial dysfunction in family amyotrophic lateral sclerosis. J Bioenerg Biomembr 43: 587-92. PMID:22072073
3. Bosco et al. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nature Neuroscience 13: 1396-1403. PMID:20953194
4. Furukawa, Y. and O'Halloran, T. (2005) Amyotrophic lateral sclerosis mutations have the greatest effect on the Apo- and reduced form of SOD1, leading to unfolding and aggregation. J Biol Chem 280 (17): 17266-74.
PMID:15691826
5. Zinman, L. and Cudkowicz, M. (2011) Emerging targets and treatments in amyotrophic lateral sclerosis. The Lancet Neurology 10 (5): 491-90. PMID:21511200
2. Faes, L. and Geert, C. (2011) Mitochondrial dysfunction in family amyotrophic lateral sclerosis. J Bioenerg Biomembr 43: 587-92. PMID:22072073
3. Bosco et al. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nature Neuroscience 13: 1396-1403. PMID:20953194
4. Furukawa, Y. and O'Halloran, T. (2005) Amyotrophic lateral sclerosis mutations have the greatest effect on the Apo- and reduced form of SOD1, leading to unfolding and aggregation. J Biol Chem 280 (17): 17266-74.
PMID:15691826
5. Zinman, L. and Cudkowicz, M. (2011) Emerging targets and treatments in amyotrophic lateral sclerosis. The Lancet Neurology 10 (5): 491-90. PMID:21511200
New to genetics? Check out this cheat sheet to learn some of the basics.


|


|
